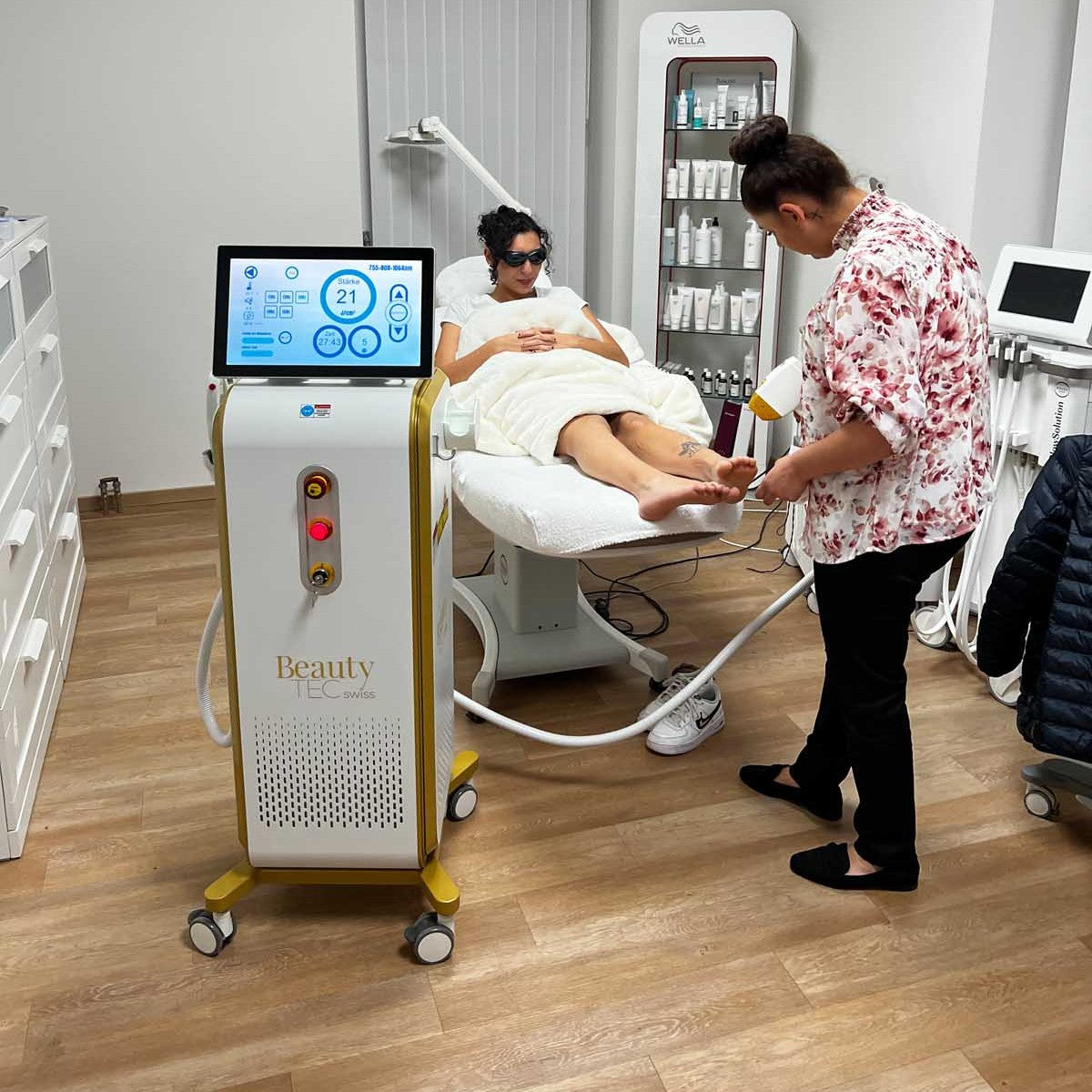
The Power of Alix Lasers


200 Million x besser !


Grüezi bei Alix Lasers ® in Zürich
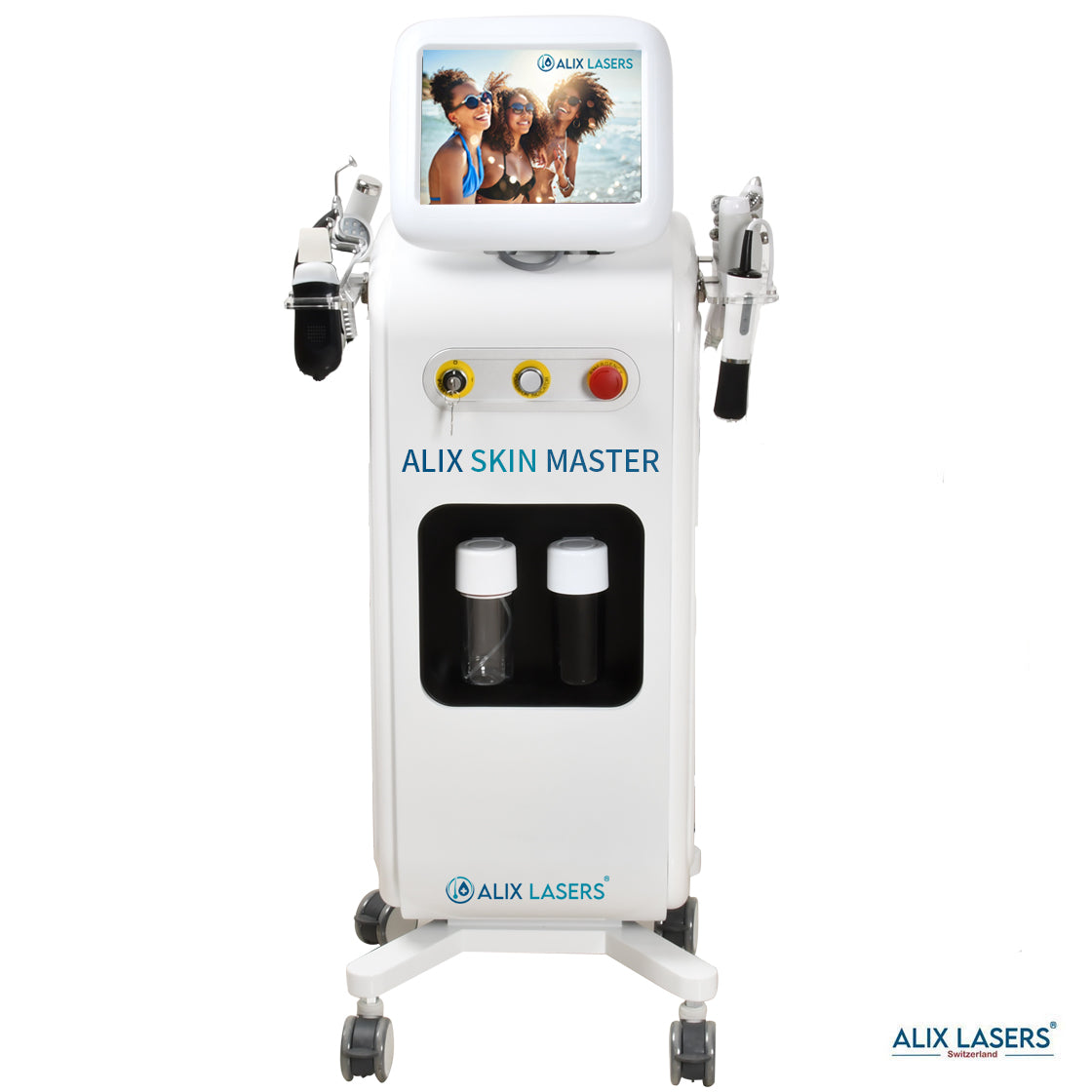






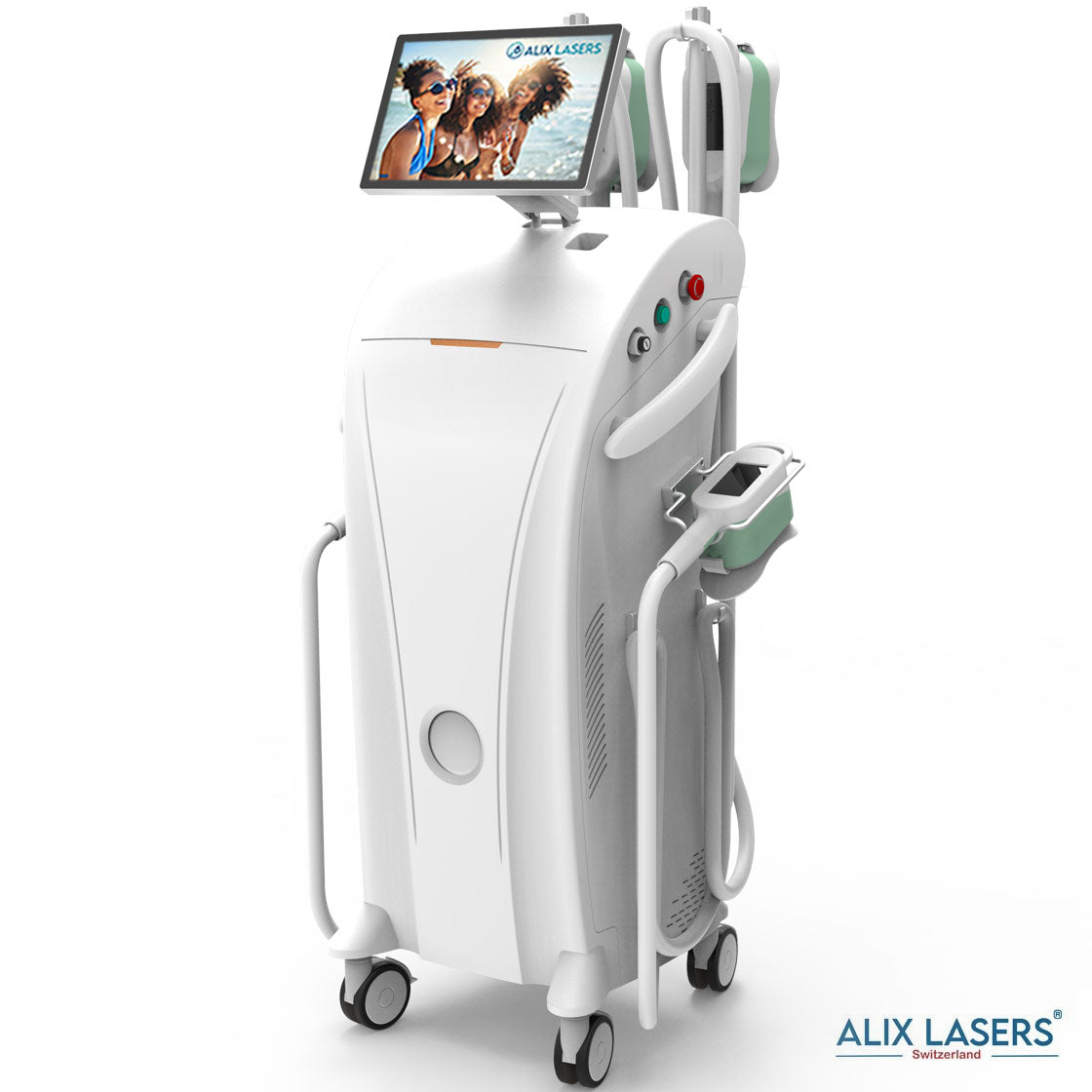
Alix Prive ® RED AI
Willkommen in der Zukunft
200 Million Schuss oder 10 Jahre Garantie

12+ Jahre OT Vision GmbH
Ihr Erfolg braucht Qualität von Alix Lasers ®
Unsere Laser- und Lichtsysteme zeichnen sich durch ihre innovative Software aus, die exklusiv an unserem Standort in der Schweiz entwickelt wird. Diese Software, die auf dem aktuellen Forschungsstand basiert, wird durch die Integration von Kundenerfahrungen ständig verfeinert. Dieser Ansatz ermöglicht es uns, eine vollautomatische Software zu bieten, die optimale Ergebnisse in allen Behandlungen erzielt.
Wir verstehen, dass die medizinische Kosmetikbranche sich ständig weiterentwickelt, und unser Engagement besteht darin, Produkte anzubieten, die nicht nur den aktuellen Anforderungen entsprechen, sondern auch auf dem neuesten Stand der Technologie sind. Dies ermöglicht es uns, unseren Kunden stets neue und wirkungsvolle Behandlungsmöglichkeiten anzubieten.
Unsere Produkte sind nicht nur am Puls der Zeit, sondern basieren auch auf höchster Technologie. Dies gewährleistet nicht nur fortschrittliche Behandlungsmöglichkeiten, sondern auch die Sicherheit und klinische Überprüfung unserer Technologien und Behandlungen. Unsere Kunden profitieren von zuverlässigen, klinisch geprüften Lösungen, die höchste Standards in der medizinischen Kosmetik setzen.
Lass Dich von uns beraten
12+
Jahre Erfahrung
26+
Länder
100%
Coherent (USA)
Schnellsuche nach Behandlung




-
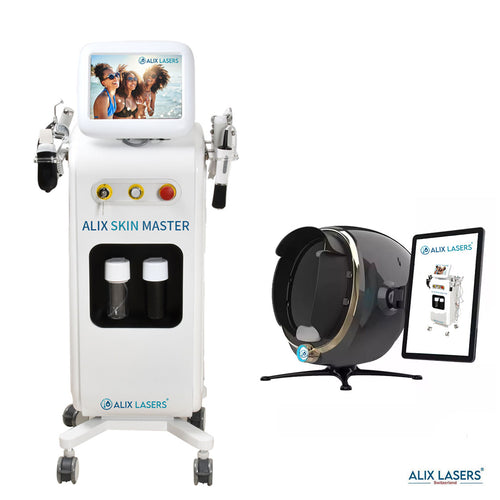
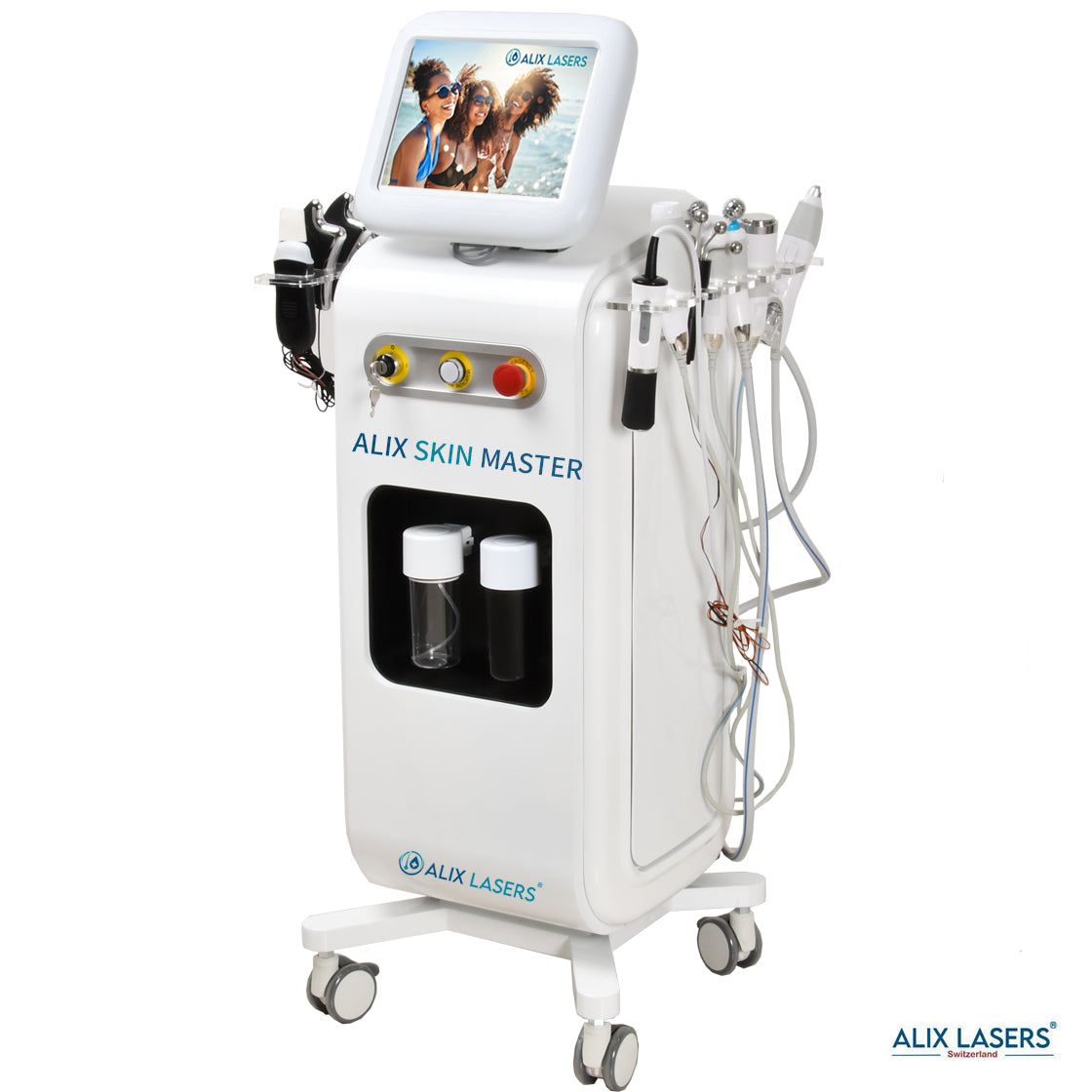
KI Intelligenz Jetzt mit Behandlungsratgeber- ALIX SKIN MASTER + FACE SCAN
-

Alix Lasers ® die neue Ära der Soforthilfe in 3D mit Apple Vision Pro
-

Alix hilft! Welches ist der beste Laser für mein Studio?
-

Alix Lasers ® präsentiert weltweit ersten KI Beauty Roboter
-

Alix Studio Onlineshop: Alles, was Ihr Kosmetikstudio braucht
-

SILENT ICE - Qualität verpflichtet seit über 12 Jahren
-

Ein Handstück für jede Zone - kinderleicht montiert und eingestellt
-

AOS 6.0 - Qualität verpflichtet seit über 12 Jahren
-

Willkommen in der ALIX SPEED ® Klasse
-
ALIX SPEED ® Ein weiterer Highlight von uns










Werde Teil der wachsenden Alix Lasers® Familie